- 关键字: 代表性成果之二:生命过程小分子调节剂的研究 发布者:宣传部 发布时间:2009-02-24 16:02:13 点击数: 19093次
|
代表性研究成果二
|
基础类或
应用基础类或基础性类
|
成果为第一完成单位
|
本室固定
人员参加
名单
|
是否
保密
|
|
生命过程小分子调节剂的研究
|
基础类
|
是
|
马大为
王任小
伍贻康
袁承业
俞 飚
|
否
|
化学遗传学是化学生物学研究最重要的组成部分。根据其研究方式的不同,化学遗传学可以分为正向化学遗传学和反向化学遗传学。正向化学遗传学是在细胞水平上筛选出能导致细胞发生表型变化的小分子化合物,然后再设法找到小分子的作用靶点,并揭示该靶蛋白的生物学功能。而反向化学遗传学是筛选出能与某个蛋白发生结合的小分子,再将筛选到的小分子作用于细胞或者生物个体上并研究其表型变化,从而了解该蛋白的功能。与经典的遗传学方法相比,化学遗传学有着可以以不同时间,不同剂量,和进行可逆操作的方式来检测特定蛋白质的功能的优点。同时它也可以在两个重要方面促进新药开发。一个方面是其鉴定出在某种疾病形成过程中起重要作用的基因或蛋白质可以被发展为新药筛选的靶点,另外一个方面是其发展的选择性地作用于某个基因或蛋白质的小分子化合物可以成为新药发现的先导化合物。因此,开展化学遗传学研究,不仅可以促进人类对于生命过程机理的了解,也可以说是进行新药源头创新的一个重要手段。因此,各国政府研究基金也对这个新兴领域给予格外的关注。如最近美国国立健康研究院(NIH)推荐的路线图计划(NIH Roadmap) 的五个研究方向中,专门设立了化学遗传学研究方向。在过去的五年里,我们在这个新兴学科,围绕“生命过程小分子调节剂的研究” 取得了一些阶段性的成绩。2004-2008期间共发表文章40余篇,这些文章已经被他人引用300多次。其中我们发展的一个趋化因子受体调节剂已经作为抗爱滋病新药完成了一期临床实验并成功实现了知识产权的国际转让,发展的细胞坏死过程小分子抑制剂也实现了知识产权的国际转让,而我们广谱的冠状病毒蛋白水解酶和细胞自吞噬的激活剂的工作分别被Nature杂志和Cell 杂志分别专文评价。具体内容介绍如下。
1.1 趋化因子受体调节剂研究
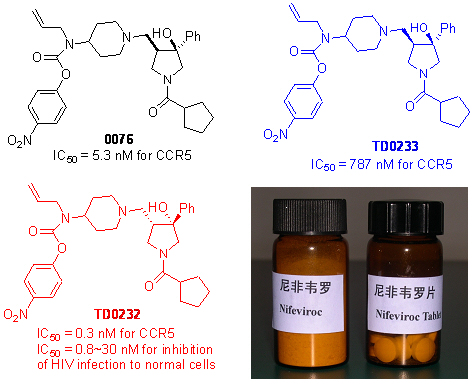
趋化因子受体是一类G-蛋白偶联受体,在免疫系统的启动方面起着决定性的作用。因此,其选择性的调节剂可以被用来治疗与免疫缺陷有关的疾病。在过去几年里,我们设计了及合成了一系列结构新颖的趋化因子受体亚型CCR5的拮抗剂,通过与上海生命科学研究院合作,发现一些化合物具有很强的对于趋化因子受体的拮抗活性(IC50<1 nM)。我们对这些化合物申请了国际专利保护(WO2005/121123,PCT/CN2008/000137),目前已经有6个国家已经批准了我们的第一个专利。通过与企业合作,我们也积极地推动在国内的药物研发工作。经过一系列临床前研究实验,我们发现其中一个化合物(TD0232,药物名字为“尼非韦罗”)不但具有很好的活性,同时也具有比较小的毒性,良好的生物利用度和药代动力学。该化合物于2006年被国家药监局批准可以作为治疗爱滋病的药物进入临床实验。目前已经完成一期临床实验,效果比较理想,计划于2009年开展二期临床实验。更值得一提的是,这些化合物的国际开发权,已经于2007年成功地转让给澳大利亚的一个专门研究爱滋病药物的公司(AVEX)。这是国内在药物开发方面实现知识产权国际转让的为数不多的例子之一,也说明我们在药物研发方面的工作得到了国际同行的认可。
1.2 冠状病毒蛋白水解酶的通用抑制剂的发现
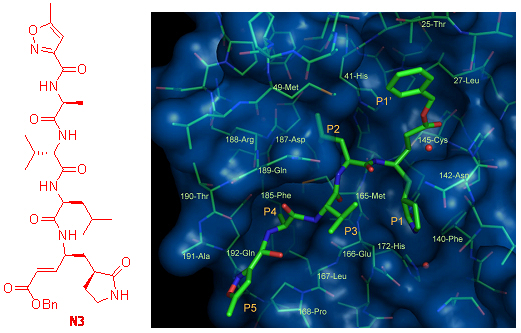
冠状病毒是一类危害人类和动物健康的病毒。研究表明,在病毒的复制过程中病毒的蛋白水解酶起到了关键的作用。因此其有效的抑制剂有可能成为治疗与冠状病毒有关的疾病。根据结构和功能的类似性,目前已知的25种冠状病毒蛋白水解酶被分为3类,属第一类的有HCoV-229E, TGEV, FIPV三种蛋白,属第二类的有MHV, SARS两种蛋白,属第三类的有IBV蛋白。我们与清华大学合作,发现一类拟肽化合物可以以很强的活性,抑制上述6种冠状病毒蛋白水解酶。这说明我们的化合物是一类冠状病毒蛋白水解酶的通用抑制剂。一些化合物(如N3)在细胞水平的实验中也体现出良好的抗病毒复制作用。通过结构生物学研究,我们发现这类拟肽化合物与冠状病毒蛋白水解酶高度保守的区域结合(见下图),这也是为什么它们具有广谱的抑制作用的原因。该工作在Plos:Biology (2005, 3, e324) 上发表以后,被Nature (2005, 437, 455), Chemical & Engineering News (Sept. 19, 2005, p9) 等杂志专文报道,认为 “这个结果为发展可以治疗由冠状病毒引起的一些疾病的药物提供了一类先导化合物”。
1.3 基质金属蛋白酶抑制剂研究
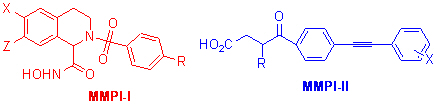
基质金属蛋白酶是一组锌依赖的蛋白水解酶。研究发现这些酶的过度表达会导致许多疾病的发生。抑制这些酶的活性的小分子调节剂有可能被用来治疗癌症等疾病。但是这个系列的酶一共有近30种,只有那些对其中的一种或者一个亚组的酶有选择性的抑制剂才有可能被发展成药物。我们与上海药物所的药理学家合作,发现了一个系列新型的拟肽类化合物(MMPI-I)有很好的抑制活性,最好的化合物的IC50为1 nM左右。与国际上活性最好的化合物的活性相当。同时发现一个系列新型的化合物(MMPI-II)对于MMP-12有特异的选择性,同时发现一个化合物可以在动物模型上以15 mg/kg剂量有效地抑制肺气肿,第一次从动物模型的角度证实了MMP-12的过度活化与肺气肿的形成有关 (J. Med. Chem. 2006, 49, 456)。
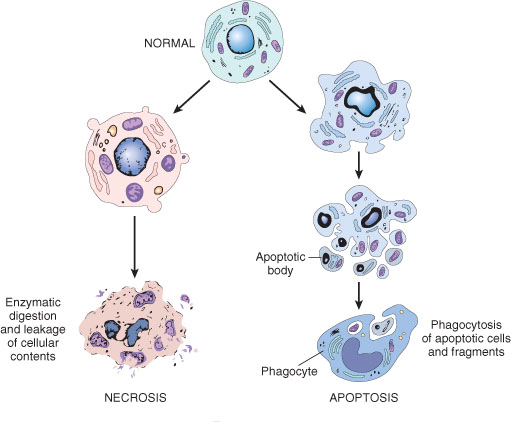
1.4 Smac蛋白模拟物及应用
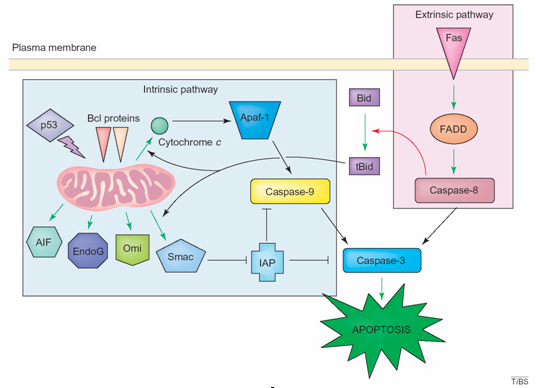
细胞凋亡 (apoptosis)是细胞接受某种信号或受到某些因素刺激后为了维持内环境稳定而发生的一种主动性消亡过程,是一种细胞的自杀性死亡。许多年以来,细胞凋亡一直是生物领域科研研究的热点。目前已知的细胞凋亡通路之一是线粒体途径(见下图),其中涉及到Smac (a second mitochondria-derived activator of caspases) 蛋白直接同细胞凋亡抑制蛋白(简称IAP)家族的XIAP,cIAP-1, cIAP-2的BIR3域以及ML-IAP的一个简单BIR域相结合,阻断XIAP对caspase-9的抑制,从而激活细胞凋亡过程。因此人工制造的外源性Smac类似物就有可能激活细胞凋亡过程,这样的分子不仅有利于促进对于细胞凋亡过程的机制研究,也有可能被用于发展新型的抗肿瘤药物。
我们根据Smac蛋白与XIAP结合的分子模建(molecular modeling)结构数据设计、合成了两个系列的构象受限的并环Smac类似物,经过活性测试我们发现有些化合物在与蛋白结合试验中显示相当出色的结合能力。其中5,8并环系列的一个化合物(DM-28)与XIAP, cIAP-1, 和c-IAP-2结合的Ki分别达到3.9, 0.37 和0.25 nM,并可以高效地抑制肿瘤细胞的生长(IC50 = 26 nM) (J. Med. Chem. 2009, 52, 593)。该化合物是目前与IAP结合活性最好的单体类Smac蛋白模拟物。目前我们正在进行这个化合物的抗肿瘤动物实验。而三并环系列中一个化合物化合物(Wu-017)与XIAP, cIAP-1, 和c-IAP-2结合的Ki分别达到18, 1.1和4.2 nM (J. Med. Chem. 2008, 51, 7352)。从测试结果我们还总结得到了此类小分子抑制剂的部分构效关系:氮甲基丙氨酸氮上甲基、脯氨酸4位芳香官能团及其立体化学以及小分子N端大位阻芳香官能团的引入对小分子抑制活性有大的贡献。

1.5 小分子自吞噬调节剂的筛选及其作用机理研究
自吞噬(macroautophagy)是一种进化上高度保守的细胞内降解过程,它与泛素-蛋白酶体降解途径(主要降解短寿命蛋白)构成两种互补的两种胞内蛋白降解机制,具有重要的生理及病理意义。例如,多种神经退行性疾病如阿尔茨海默病、亨廷顿舞蹈病的发生均源于细胞中错误折叠蛋白聚合沉积,后者产生细胞毒性导致神经元功能失常。因此,诱导自吞噬可能成为神经退行性疾病治疗的新靶点。而目前常用的自吞噬诱导剂rapamycin虽然被证明可以促进错误折叠蛋白降解,但其较高的毒性以及不能通过血脑屏障等都使之无法应用于神经退行性疾病治疗。因而,利用化学生物学的手段寻找新的自吞噬诱导剂意义重大。另外,目前研究自吞噬的分子工具也并不十分理想,常用的两类自吞噬抑制剂如氯喹、巴弗洛霉素(通过破坏溶酶体的结构和功能阻止自噬性降解)以及3-甲基腺嘌呤(3-MA)、渥曼青霉素(抑制磷脂酰肌醇3-磷酸激酶(PI3K)活性)均存在效率低、毒副作用大等缺陷。这些空缺急需化学生物学研究者加以填补。
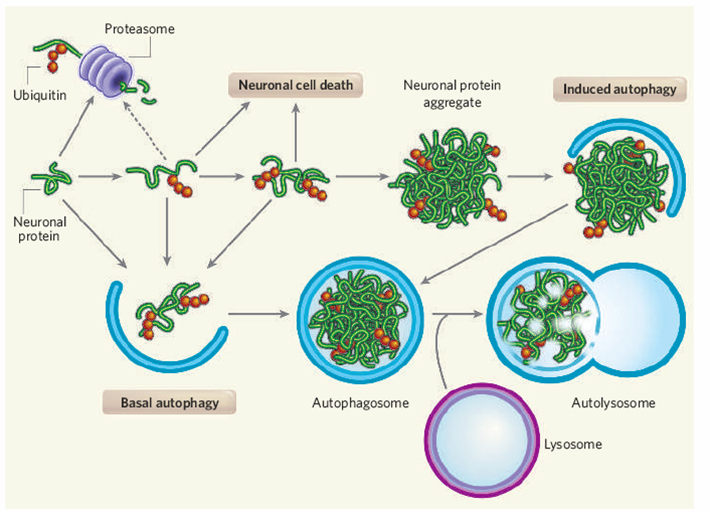
几种新的小分子自吞噬诱导剂的发现:我们利用自己建立的H4-LC3细胞模型,通过高内涵筛选筛选了几千种已知和未知生物活性的化合物。对于初筛中发现的有苗头的化合物,我们进一步建立H4-FYVE细胞筛选模型并结合长寿命蛋白降解、错误折叠蛋白降解等方法进一步确证,最终发现了8种新的自吞噬诱导剂。其中7种为FDA业已批准的药物。氟司必林、三氟拉嗪以及匹莫齐特是FDA批准的用于治疗精神分裂症的镇静剂。另外3种自吞噬诱导剂尼古地平、尼卡地平以及胺碘酮则是FDA批准的治疗心血管疾病,包括高血压、心绞痛、心率不齐等的药物。以上化合物都能够通过血脑屏障。洛哌丁胺则是FDA批准的治疗痢疾的药物,它可以有效改善胃肠道症状。青霉震颤素是一种麦黑草(ryegrass)中发现的真菌神经毒素, 可以选择性阻断Ca2+ 激活的K+ 通路(10 nM青霉震颤素即可100% 阻滞)。而此次新发现这些化合物具有自吞噬诱导的新活性,扩展了这些化合物(药物)的用途,可能实现“老药新用”,可望被用来治疗与自吞噬有关的一些神经退行性疾病的治疗。该工作在Proc. Natl. Acad. Sci. USA. (2007, 104, 19023)上发表后被国内外多个科研网站报道并被 Cell 杂志选入Leading Edge/Cell Biology Select (Cell, 2007, 131, 1211) 给予高度评价,认为“该工作为分析了解自体吞噬的机制以及亨廷顿舞蹈病等神经退行性疾病的治疗提供了重要资料”。
新型小分子自吞噬抑制剂的研究:通过筛选,我们在抑制剂的研究方面亦有很大收获,首先,我们筛选到了一个高效抑制自吞噬的化合物4E3,该化合物对自吞噬的抑制作用具有高效性(EC50=0.788μM,而目前常用的autophagy抑制剂3-MA的工作浓度为10 mM)、低毒性(未见细胞损伤或周期阻滞)和普适性(抑制rapamycin或饥饿等多种因素诱导的自吞噬,对多种细胞系均有作用)等特点。机理研究显示该化合物通过影响自吞噬体形成过程中重要的蛋白复合体Vps34/Beclin1复合物形成有关。也就是说,该化合物仅影响自吞噬体形成,并不影响多种通路上的重要蛋白(如PI3K)活性或其它细胞器(例如溶酶体)功能,因而高效专一,毒副作用很低。我们在此基础上进一步设计并合成了100余种衍生物加以筛选,发现一个化合物C29的活性提高了近10倍(EC50=0.084μM),而一些化合物(如C43)活性与4E3相当,但去除了4E3原有的PDE5抑制剂活性(该活性已经证明与化合物诱导自吞噬的新活性无关),成功实现了活性拆分,因而具有更好的选择性。以上工作为研究自吞噬提供了有用的“工具药物”。
1.6 小分子高尔基体干扰剂的筛选及机理研究
一直以来,针对高尔基体的研究始终是细胞生物学研究的一个热点和难点。高尔基体的主要功能是将内质网合成的多种蛋白质进行加工、分类与包装,然后分门别类地运送到细胞特定的部位或分泌到细胞外。然而这些过程如何发生、蛋白如何被识别加工等具体细节尚不清楚。对此,我们也也建立了两种荧光标记高尔基体的细胞模型H4-p58(标记顺面高尔基体)以及H4-GalT细胞(标记反面高尔基体,试图利用化学生物学手段高通量筛选特异作用于高尔基体的小分子。从高尔基体形态来看,在筛选过程中,高尔基体可能发生两类变化:一是荧光标记高尔基体形态完好,并且增大变亮。高尔基体体积增大可能意味着高尔基体所执行的蛋白分泌功能旺盛;另一类变化则是高尔基体分散,而在目前研究中,人们常常用各种小分子高尔基体干扰剂来干扰和破坏高尔基体,使其处于特定的分散状态,进而研究和调控高尔基体的各种功能。我们也通过分别筛选到了促分泌的小分子或干扰高尔基体功能的小分子。
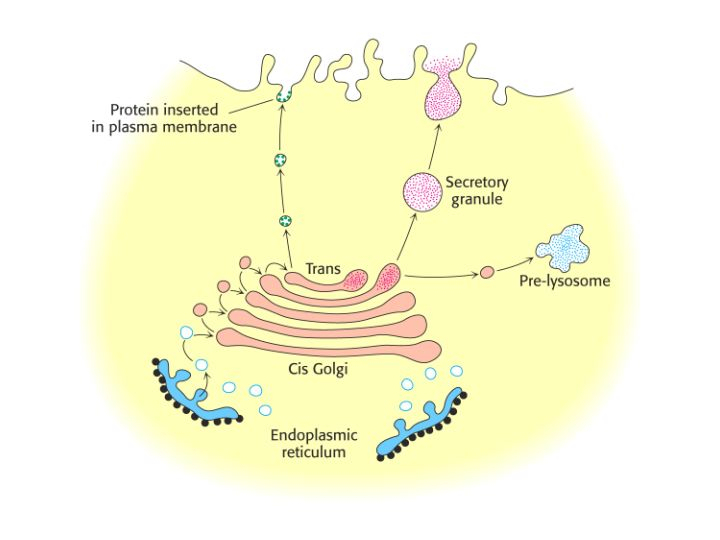
促进高尔基体分泌的小分子sioc145:在筛选中我们发现,一些化合物可以导致高尔基体变大变亮。这些化合物包括本研究室之前合成的几种活性分子sioc109、145、159、162等,一系列内源高尔基体蛋白的免疫荧光以及电镜观察均显示sioc145促进高尔基体膨大的效果非常明显。因此我们选取sioc145作为研究对象,并以另一种结构相似但无活性的化合物sioc111作为阴性对照。通过一系列细胞活性试验以及建立分泌性碱性磷酸酶表达的细胞模型进一步研究,我们发现该化合物能够促进细胞代谢旺盛,促进高尔基体分泌功能。进一步的细胞实验研究我们发现该化合物可以促进胰岛素由胰导细胞分泌,从而直接调控体内血糖水平。这说明该化合物可以抑制糖尿病的发生。通过动物实验我们已经初步证明了这个设想。目前正在深入研究阶段,希望使这类化合物成为作用机制新颖,具有自主知识产权的药物候选分子。
小分子高尔基体干扰剂AG1478及其作用机制: 除sioc145这类促分泌剂之外,我们筛选到的另外一类影响高尔基体的化合物则是能够选择性分散高尔基体的工具分子。AG1478即能够选择性地破坏高尔基体,使得高尔基体很快分散于胞质中,其效果显著(EC50 = 3.35 M)、作用迅速(t1/2 ≈ 10 min)、可逆。虽然AG1478之前被认为是一种已知的高选择性的EGFR抑制剂(文献报道其对EGFR的抑制浓度约为500nM),但我们通过用多种已知的EGFR抑制剂比较以及RNA干涉EGFR证明AG1478分散高尔基体的作用与EGFR无关,属于影响新的靶点体现出的新活性。通过与几种能够分散高尔基体的化合物比对,我们发现AG1478分散高尔基体并非通过影响细胞内pH、ATP水平及细胞骨架来实现,而是类似于目前广泛使用的小分子高尔基体干扰剂BFA,是通过灭活重要高尔基体蛋白Arf1来分散高尔基体。但AG1478并非BFA活性的简单重复,其作用机理与BFA并不相同,首先,BFA对顺式,反式高尔基体蛋白均能够很好分散,而AG1478只对顺式高尔基体蛋白作用明显,体现出分散顺、反式高尔基体蛋白的选择性。其次,该活性仅仅体现于人源的细胞系而对鼠源细胞系没有作用,也不能促进鼠源Arf1失活。通过对Arf1上游蛋白GBF1的研究,我们发现AG1478可以改变hGBF1在细胞中分布,从而影响Arf1活性。其作用位点位于GBF1蛋白N端DCB domain。而BFA则作用于GBF1蛋白Sec7 domain。BFA的作用可以将hGBF1蛋白直接运送回内质网,而AG1478作用并未将hGBF1蛋白运送回内质网,而是聚集于内质网出口附近。并且与BFA比较,AG1478毒性更低,效果更加专一,是一种更加细化地研究高尔基体的工具分子。结果发表在 J. Biol. Chem. (2008, 283, 31087)。

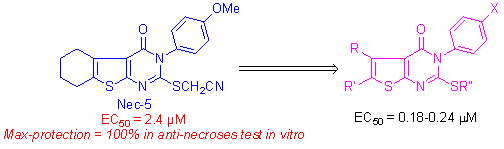
1.7 Necroptosis过程小分子抑制剂的研究:
细胞是组成有机体的单元结构,因此通过小分子对细胞程序性凋亡或非程序性坏死信号传递过程的考察可视为创制新药新途径的基础研究。最近袁钧瑛等的研究揭示存在着一种不依赖caspase的、形态特征上类似细胞坏死的程序性细胞死亡,这种新的细胞死亡方式命名为necroptosis,并找到一种化学小分子necrostatin-1(Nec-1)能专一性的阻断FasL/TNFα诱导的细胞坏死,但对细胞凋亡没有抑制作用,证明了确实存在着一种由精确的细胞信号通路来引起细胞坏死的代谢途径。动物实验进一步证明Nec-1减少了缺血性脑伤害的程度。细胞坏死现象广泛存在于缺血性损伤(如中风和心肌梗塞)、创伤和一些急性和慢性神经退化性疾病(如老年痴呆症)等疾病。由此可见,necrotptosis的研究为预防和治疗中风等一类疾病开阔了新的思路。因此,设计与研究影响细胞necroptosis有机小分子的合成、结构及构-效关系实质上是考察这些小分子探针的信号传递过程,是寻找新药的有效途径。可能发展成为研制新药的新方法,我们与哈佛医学院的袁钧瑛合作,对四大类necrostains即Nec-5,Nec-7,Nec-8及Nec-12的化学结构进行修饰并研究其结构与活性关系,发现了活性提高了10几倍的化合物。同时对上述四大类高活性化合物进行链接化学(click chemistry)与biotinylation化研究,以考察小分子与细胞结合的靶点。部分结果已经在Nature Chemical Biology上发表(其中有机所为参与单位)。基于这个结果,美国哈佛大学和上海有机所等也联合申请了专利进行知识产权保护。值得一提的是,这个专利的知识产权于2008年成功转让给美国的TetraLogic公司。该公司将投入人力和物力,以进一步把我们的先导化合物发展为治疗神经性疾病的药物。
1.8 基于生物靶标分子结构的药物设计方法研究
基于生物靶标结构的药物设计(structure-based drug design)是合理设计能够调控给定生物靶标功能的有机小分子活性化合物的有力工具。在逆向化学生物学研究中应用该类方法,有可能合成和实验测定数目较少的化合物就得到活性较高的化合物。尤其当待研究的生物靶标尚缺乏已知的有机小分子调控剂作为参照时,该类方法就可以发挥更大的作用。在过去的几年中,我们致力于发展根据生物靶标进行配体分子合理设计的程序方法以及对药物分子的关键性质进行评估的计算方法,为药物设计提供性能更加可靠、功能更加完备的工具方法。
a) 我们发展了识别有机化合物化学结构的I-interpret算法(J. Chem. Inf. Model. 2007, 47, 1379)。该算法可以根据有机化合物最基本的结构信息(包括原子类型以及坐标)来自动识别出化合物的正确化学结构,可用于在虚拟筛选中对大型有机化合物数据库的预处理。通过对NCI Diversity Set中1990种类药有机分子的测试,I-interpret方法对化学结构识别的准确度为98-100%,显著超过了目前流行的Babel等算法。
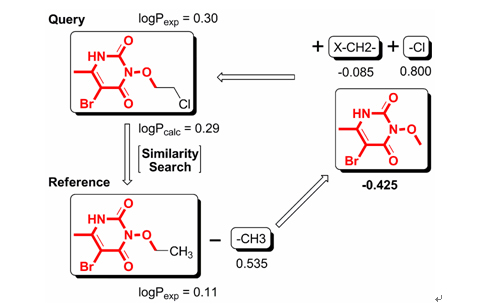
b) 我们发展了用于预测有机化合物脂水分配系数(logP)的XLOGP3方法(J. Chem. Inf. Model. 2007, 47, 2140-2148),用于在虚拟筛选中应用类药性判定规则过滤不符合要求的化合物。该算法采用了加合法模型结合结构相似性搜索的新策略,可以利用不断增长的实验数据有效地提高精度。最近Pfizer公司利用其内部超过96000种化合物的logP实验数据对当今30余种logP算法进行了对比评估(J. Pharm. Sci. 2008, 98, 861),结果显示XLOGP3在各项测试中均名列前茅,是当今世界上最准确最有效的logP算法之一。
c) 为了配合对蛋白-小分子配体亲合性算法的研究,我们构建了PDBbind-CN数据库。它是世界上第一个系统地收集Protein Data Bank数据库中各类复合物(蛋白-小分子配体、核酸-小分子配体、蛋白-蛋白以及蛋白-核酸复合物)亲合性实验数据的数据库。该数据库可以通过网络来访问(http:// www.pdbbind.org.cn/),并提供对数据的分析检索以及分子结构的显示等功能。自2007年底公开发布以来在线总访问量已经超过23000余次,并被世界上多个文献检索机构所推荐。该数据库目前得到定期更新,所包含的数据量以每年约20%的速度在增长。
自2007年以来,我们所开发的药物设计方法和数据库已获得国家计算机软件著作权共计5项。其中I-interpret软件、XLOGP3软件以及PDBbind-CN数据库已先后公开发布,引起了国内外同行的广泛关注。目前共计拥有分布在四十余个国家的超过1400名注册用户,其中许多来自国际上著名的大学和研究机构(如NIH、Harvard、MIT等)以及大型制药公司(如Pfizer、Abbott、Merck等)。


 沪ICP备05005485号
沪ICP备05005485号