- 关键字: 有机合成新反应和新方法的研究及其在复杂生物活性分子合成中的应用 发布者:宣传部 发布时间:2009-02-24 22:02:58 点击数: 9739次
|
代表性研究成果五
|
基础类或
应用基础类或基础性类
|
成果为第一完成单位
|
本室固定
人员参加
名单
|
是否
保密
|
|
有机合成新反应和新方法的研究及其在复杂生物活性分子合成中的应用
|
应用基础类
|
是
|
马大为
俞飚
袁承业
伍贻康
姚祝军
|
否
|
如前所述,化学生物学研究的一个关键环节是对于具有重要生物活性的天然产物或选择性结合生物大分子的小分子调节剂的发现。生物大分子的多样性要求能与之选择性结合的小分子化合物具有广泛的结构多样性。为了高效地创造多样性的有机小分子,化学家需要针对其中的特殊骨架、连接、和官能团等发展更多新的合成反应和合成方法。新的反应有可能彻底革新合成的策略和途径,近来最重要的例子如Sharpless开创的不对称环氧化、双羟化和羟胺化(2001年诺贝尔化学奖)、Grubbs等发展的烯烃复分解反应(2005年诺贝尔化学奖)和各类碳-碳和碳-杂原子偶联反应等。
在过去五年中,我们针对一些复杂生物活性分子,如多类生物碱、杂环、糖缀合物、多醚类化合物和手性氨基膦酸等的合成,发展了一系列新反应和新方法,如氨基酸促进的Ullmann类反应、有机小分子催化的不对称Michael反应、糖苷化反应和不对称Aldol反应等。这些工作总共发表论文82篇,其中影响因子大于5的11篇;这些论文的他引总数已经达到800余次。这里的大部分工作曾作为主要内容,分别获得了国家自然科学二等奖(2007)、上海市科技进步一等奖(2005)和上海市自然科学二等奖(2006)。其中,受到较大关注的论文包括:
|
J. Org. Chem. 2005, 70, 5164
|
已经被他引143次;
入选2006年ACS活动周中评选出的当时被引用最多的100篇化学论文;
在2008-2009年度被J. Org. Chem.杂志评选为最近3年来该杂志发表的所有文章中被引用最多的10篇文章之一。
|
|
Org. Lett. 2005, 7, 5545
|
被国际网络杂志Organic Chemistry Highlights亮点介绍。
|
|
J. Am. Chem. Soc. 2006, 128, 16050
|
被Angew. Chem. Int. Ed. 在其Highlights栏目以及国际网络杂志Organic Chemistry Highlights亮点介绍。
|
|
Angew. Chem. Int. Ed. 2006, 45, 1276
|
被该期刊列为hot paper。
|
|
Angew. Chem. Int. Ed. 2007, 46, 2598
|
被该期刊列为hot paper;
被Organic Process Research & Development 在其Highlights from the Literature栏目中介绍。
|
|
J. Am. Chem. Soc. 2007, 129, 9300
|
被国际网络杂志Organic Chemistry Highlights亮点介绍。
|
|
Acc. Chem. Res. 2008, 41, 1450
|
Cross-Coupling专辑特邀论文。
|
值得一提的是:我们2006-2008阶段发展的氨基酸促进的Ullmann反应在发表以后,已在他人的合成研究中得到60多次成功应用,用于合成设计的生物活性、材料分子以及发展新的合成方法等(这些证明材料见附件5)
以下是所取得的代表性重要进展:
1. 氨基酸促进的Ullmann类反应
芳基卤代物、烯基卤代物与亲核试剂的偶联是有机合成中的基本反应。发展条件温和、使用廉价催化体系的新反应是有机化学学术界和工业界追逐的目标。在我们对于氨基酸促进的Ullmann反应研究的基础上,在过去的五年里在这个方向上又取得了一系列的成果。例如,我们发现氨基酸作为促进剂也可以使含氮杂环的芳基化反应、酰胺与烯基卤代物的偶联反应以及取代亚磺酸钠与芳基卤代物的偶联反应在相对温和的条件下进行。在此基础上,我们发展了芳基叠氮化物的合成新方法和不用钯催化剂的末端炔与芳基卤代物偶联的Sonogashira反应的新条件。发现在氨基酸促进下,芳基卤代物与活泼亚甲基类化合物的偶联反应可以在室温到50oC下进行。利用这些我们自己发展的酰胺与烯基卤代物的偶联反应,我们完成了环肽类生物碱ziziphine N,Abyssenin B和mucronine E的首次全合成。
.jpg)
.jpg)
.jpg)
同时,我们也发现了芳基卤代物的邻位酰胺基团对于Ullmann反应有邻基促进效应,结合这个效应和氨基酸促进的效应,我们发展了室温下进行的芳基卤代物与苯酚进行偶联反应而生成二芳醚的反应。这是Ullmann类反应目前为止最温和的反应条件。这个结果对于合成许多具有二芳醚骨架的,具有抗菌和抗肿瘤等活性的环肽化合物具有重要意义。这个工作作为Angew. Chem. Int. Ed. (2006, 45, 1276) 的hot paper发表。
通过进一步研究,我们也发现了可以使一般的带有氨基酸单元的芳基卤代物和苯酚的偶联反应在不消旋的条件下进行。利用这些结果,我们发展了目前最有效的合成天然产物K-13和L,L-isodityrosine的方法。
.jpg)
结合邻位酰胺基团对于Ullmann反应有邻基促进效应和氨基酸的配体效应,我们发现在-45oC下芳基卤代物与取代的活泼亚甲基类化合物的偶联反应就可以进行,这是目前Ullmann反应最低的反应温度。进一步研究发现利用光学纯的氨基酸为配体,可以进行不对称的C-C键形成反应,ee最高可以达到93% (J. Am. Chem. Soc. 2006, 128, 16050)。这项工作被Angew. Chem. Int. Ed. (2008, 47, 3096) 在其Highlights栏目专文介绍,同时也被国际网络杂志Organic Chemistry Highlights (http://www.organic-chemistry.org/Highlights/2007/23July.shtm) 亮点介绍。我们还发现带有邻位酰胺基团的芳基卤代物与活泼亚甲基类化合物的偶联反应所得到的产物可以分别转化为取代吲哚和氧化吲哚。带有邻位酰胺基团的芳基卤代物与胺的偶联反应可以得到多取代的苯并咪唑,而带有邻位碳酰胺基团的芳基卤代物与胺的偶联反应可以得到多取代的苯并咪唑酮。该工作在Angew. Chem. Int. Ed. (2007, 46, 2598) 作为hot paper发表。并被Organic Process Research & Development在其 Highlights from the Literature栏目中介绍。
.jpg)
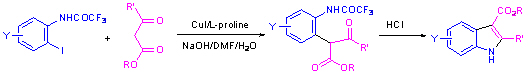
.jpg)
除了上述的杂环合成的新方法外,在氨基酸促进的Ullmann类反应研究的基础上,我们也发展了合成其它杂环如苯并呋喃、2-三氟甲基吲哚、3-酰基氧化吲哚、多取代异喹啉以及一些三并杂环等。这些杂环体系广泛存在于已知的药物和生物活性分子中。我们发展的合成新方法与已知的方法相比大部分都有操作简单和普适性强的特点。因此,在杂环合成中将有较好的应用前景。
.jpg)
上述有关氨基酸促进的Ullmann类反应的工作已被Acc. Chem. Res. 邀请撰写了评论性文章,被Synthesis和Synlett杂志邀请在其“实用合成方法” 和“Cluster”栏目中撰文介绍。部分工作也分别被Angew. Chem. Int. Ed. 在其Highlights栏目、Organic Process Research & Development在其 Highlights from the Literature栏目、以及国际网络杂志Organic Chemistry Highlights 等专文介绍。多篇文章被评为most cited 或most access papers。例如,我们2005年的J. Org. Chem.(2005, 70, 5164)文章入选2006年ACS活动周中评选出的当时被引用最多的100篇化学论文,并在2008-2009年度被J. Org. Chem.杂志评选为最近3年来该杂志发表的所有文章中被引用最多的10篇文章之一。更值得一提的是我们在2004-2008期间发展的这些合成方法在过去几年里已被他人成功应用67次,使得我们在氨基酸促进的Ullmann类反应的工作的他人应用达到100余次。这些应用主要是用来合成相关的生物活性分子、天然产物、杂环及杂原子化合物。
2. 吡啶烷、Indolizidine和Quinolizidine类生物碱的合成新方法
吡啶烷、Indolizidine和Quinolizidine类生物碱及相应的杂环骨架不仅广泛存在于天然产物中,而且在人造的生物活性分子中也非常常见。因此,对于它们的高效、不对称的合成研究一直是有机化学的热门课题。我们在以前利用-氨基酸衍生物作为手性源合成生物碱工作的基础上,在过去五年里又发展了一些高效的串联反应的过程来构筑吡啶烷、Indolizidine和Quinolizidine类生物碱的新方法。它们包括:(1) 以-氨基氯丙烷与炔基酯或炔基酮的Michael加成/取代反应为串联反应过程的合成吡啶烷、Indolizidine和Quinolizidine类生物碱的方法 (J. Org. Chem. 2005, 70, 7364)。(2) 以炔基酯的碘代物与氮杂环丙烷的的取代反应/表观[3+2]环加成反应为串联反应过程的合成Indolizidine类生物碱的方法 (Org. Lett. 2005, 7, 5545)。这项工作被国际网络杂志Organic Chemistry Highlights (http://www.organic-chemistry.org/Highlights/2006/24April.shtm) 亮点介绍。(3) 炔烃对于活化吡啶的不对称催化Michael加成。这是第一例不对称催化的对于简单吡啶的碳-碳键形成反应,具有较高的合成应用价值。例如,利用这个反应我们发展了对于indolizidine 223AB和 indolizidines 167B的目前最短的合成路线 (J. Am. Chem. Soc. 2007, 129, 9300)。这项工作也被国际网络杂志Organic Chemistry Highlights (http://www.organic-chemistry.org/Highlights/2008/12May.shtm) 亮点介绍。
.jpg)
3. 有机小分子催化的不对称Michael反应
有机小分子催化下醛对硝基烯的Michael加成反应最近得到了很多关注,主要是这个方法可以以经济的方法提供一些手性有机合成中间体。但是,以前的方法均存在需要使用过量的醛、比较高的催化剂用量、反应速度慢、以及对于一些底物的对映和非对映选择性不理想等问题。我们发现用二苯基脯氨醇硅醚为催化剂,苯甲酸为添加剂,水为反应介质的反应体系可以在根本上解决这些问题 (Angew. Chem. Int. Ed. 2008, 47, 545)。这一发现大大提高了这个反应的实用性。从机理上来讲,这个反应的成功得益于催化剂Diarylprolinol Ethers的良好的不对称诱导能力、苯甲酸促进其与底物醛快速的烯胺的形成、以及水所起的作用。接下来,我们将这个催化体系用于醛对于-酮基-,-不饱和酯的Michael加成和进一步环化上,发展了一个不对称合成多取代的6元环内酯的新方法 (Org. Lett. 2008, 10, 3075)。同时,我们也发现在二苯基脯氨醇硅醚催化下醛可以和-酮基-,-不饱和酯进行Michael加成,生成的产物经过简单的转化就可以给出多取代的环己烯酮、环己酮、吡啶烷和-丁内酯等结构 (Org. Lett. 2008, 10, 5425)。这些方法有望在合成具有重要生物活性的复杂分子中得到应用。
.jpg)
4. 新型糖苷化反应
糖苷化反应是寡糖和糖缀合物合成中的关键反应。我们进一步研究了我们在此前发展的以糖基三氟乙酰亚胺酯为糖基给体的新的糖苷化方法,并在一些具有良好生理活性的复杂糖缀合物的合成中得到了很好的应用,如使用该方法完成了对具有抗肿瘤活性的常见植物呋甾皂甙Protodioscin (Org. Lett. 2006, 8, 2679)、具有抗菌作用的海绵糖脂Caminoside A (Synlett 2005, 437)、第一个被发现的具有开环糖连接的天然糖缀合物Anemoclemoside B (Org. Lett. 2005, 7, 1935)、和人参皂甙Ro (Synlett 2004, 259)等的全合成。证明了该方法具有通用性和高效性。
.jpg)
.jpg)
这一方法克服了以往最常用的Schmidt糖苷化法中存在的问题,如活泼给体的制备和分子内转糖苷化等,得到了国际同行的广泛关注和应用。例如:日本的Takahashi小组用于糖肽的合成和抗生素的固相合成;日本的Fukase小组、美国的Huang小组和国内的Li小组用于含唾液酸糖缀合物的合成;法国的Papot小组用于hydroxyamic acid的直接糖苷化;瑞士的Seeberger小组和美国的Boons小组用于复杂糖脂的合成和固相合成;美国的Crich小组用于氟代糖的糖苷化;意大利的Adinofi小组、Bedini小组、俄罗斯的Nifantiev小组等用于活泼糖基的糖苷化等等。关于该反应的最早2篇论文(J. Org. Chem. 2002, 67, 9099; Tetrahedron Lett. 2001, 42, 2405)已被他人引用80余次。
此外,我们还发展了Au(I)催化的1,2-内醚糖的糖苷化反应(J. Org. Chem. 2008, 73, 4323)。该反应能直接获得2位羟基裸露的1,2-反式糖苷化合物,该2位羟基则可以直接用于下一步的糖苷化反应。与传统的ZnCl2促进的反应相比,新反应的产率提高了20%以上。
传统的糖苷化反应大都在酸性条件下进行,并需使用当量的促进剂。在对复杂分子的合成中,酸性条件可能引起副反应。理想的糖苷化反应应该是在催化量的促进剂作用下在中性条件下进行。我们发展了以邻-炔基苯甲酰基为离去基的新型糖苷化反应(Tetrahedron Lett. 2008, 49, 5036)。该类糖基给体能够稳定存放,易于制备。反应在金(I)的催化下,在中性条件下进行,产率高,适用底物广。有望发展成一个应用广的高效糖苷化反应。
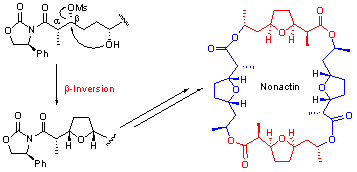
5. 基于aldol -位翻转反应的合成-内酯和四氢呋喃环新方法
-内酯和四氢呋喃环都是天然产物中常见的结构片段。Lipstatin及其衍生物tetrahydrolipstatin (即已上市的减肥药“奥立司达”Orlistat)之类的化合物都含有一个trans双取代的-内酯环作为关键的药效部分。采用经典的Adams条件(PhSO2Cl/Py)来合成这种-内酯环需要用光学anti aldol(远不如相应的光学活性的syn aldol容易合成)作为前体,是的合成变的复杂。我们发展了新的-位构型翻转的条件,是的可以用直接利用Evans aldolization的产物syn aldol, 在活化了-位羟基后用羧基作为进攻基团,得到-位构型翻转的内酯 (Chem Comm 2005, 1906)。
.jpg)
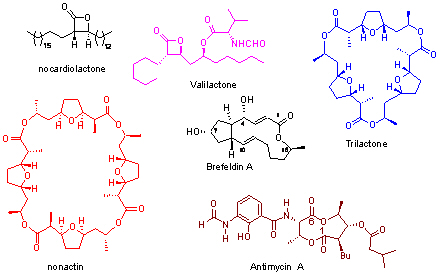
当进攻基团是分子内相隔2个CH2单元的一个羟基时,还可以形成四氢呋喃环;为合成Ionophore类的抗生素Nonactin和Trilactonen等提供了简洁的合成途径:(Org. Lett. 2006, 8, 2831)
基于上述这些方法研究的结果我们还通过高效的新路线成功地合成了具抗真菌/抗肿瘤活性的Brefeldin A (J. Org. Chem. 2004, 69, 3857; Org. Lett. 2008, 10, 2831) 和Antimycin A (J. Org. Chem. 2006, 71, 4296), 脂酶高效抑制剂Valilactone (IC50 = 0.35 nM,比临床上用的Orlistat活性高3个数量级)(J. Org. Chem. 2006, 71, 5748), 以及抗菌的天然内酯Nocardiolactone等天然产物,并通过全合成推定了天然Nocardiolactone的绝对构型(Synlett 2005, 1477)。
6. 合成抗流感药物达菲和乐感清的新路线
唾液酸是广泛存在于生物系统中的一类天然糖类化合物,在细胞膜和神经组织中含量很高,大多通过2-位异头碳的羟基以-糖苷键连接在糖蛋白、糖脂和寡糖的末端,因此被人们称为多糖类化合物“天线”,在许多重要的生理过程中起关键的作用。通过衍生物结构的变化研究唾液酸在细胞过程中的作用,对细胞表面进行化学修饰,对于探索细胞代谢产物和重要蛋白质的糖苷化等均有重要意义,也是全球性的热点科学问题。同时,唾液酸也是著名的流感药物达菲(Tamiflu,现由莽草酸为原料生产)和乐感清(Relenza,现由唾液酸为原料生产)的结构设计原形。
.jpg)
从葡萄糖酸内酯等简单且来源丰富的原料出发,我们在过去的几年里发展了通过针对性的非对映选择性的加成反应条件、化学选择性氧化反应等成功实现了一条乐感清合成的新路线。个别唾液酸类中间体还在相应的细胞生物学和代谢研究中获得新的应用(Org. Lett. 2004, 6, 2269);通过研究Grubbs催化剂介导的烯烃复分解反应条件(J. Org. Chem. 2004, 69, 5314; Tetrahedron Lett. 2005, 46, 8567),实现了从来源丰富的丝基酸出发的达菲骨架和全部官能团的建立方法 (J. Org. Chem. 2006, 71, 5365)。这些方法探索和合成应用工作对于今后发展流感药物新的生产路线以及合成具有多官能团的环己烷类化合物具有较好的参考价值和借鉴作用。
7. 合成抗癌天然产物喜树碱类化合物的串联反应新路线
喜树碱 (camptothecin)是从我国喜树中分离得到的拓扑异构酶I (Topo I)的特异抑制剂,能有效抑制DNA的合成。喜树碱及其类似物对30余种肿瘤都有不同程度的疗效,是继红豆杉之后第二个重要的木本抗癌药用植物。目前除了已经上市的喜树碱类抗癌药Topotecan (Hycamtin)和Irinotecan (Camptosar)外,还有Kaarenitecin等多个喜树碱类化合物处于临床阶段。但是我国喜树的野生资源已经处于濒危状态,在某些地方已经绝迹,现存的少量的野生资源也存在灭绝的危险;目前全世界的喜树碱的生产能力仅为600 Kg。显然,喜树碱资源远远不能满足市场的需要,利用化学合成的方法大量获得喜树碱及其类似物成为亟待解决的问题。
过去几年中,我们发展了一些高效率的串联反应快速构建喜树碱典型骨架,先后完成了两条全新的喜树碱及其衍生物的全合成路线(Org. Lett. 2007, 9, 2003; Org. Lett. 2008, 10, 5393; 中国发明专利200810042106.2)。这些新发展的路线具有原料易得、路线短、操作简单、条件温和和总产率高等特点,并可以延伸到喜树碱衍生物的合成以及用于其它具有喜树碱类似结构的天然生物碱的全合成中(J. Org. Chem. 2007, 72, 6270)。


 沪ICP备05005485号
沪ICP备05005485号